|
SSAGES
0.1
A MetaDynamics Package
|
|
SSAGES
0.1
A MetaDynamics Package
|
Collective variable to measure antiparallel beta secondary structure. More...
#include <AntiBetaRMSDCV.h>
Public Member Functions | |
| AntiBetaRMSDCV (std::vector< int > resids, std::string refpdb, double unitconv, int mode) | |
| Constructor. More... | |
| void | Initialize (const Snapshot &snapshot) override |
| Initialize necessary variables. More... | |
| void | Evaluate (const Snapshot &snapshot) override |
| Evaluate the CV. More... | |
 Public Member Functions inherited from SSAGES::CollectiveVariable Public Member Functions inherited from SSAGES::CollectiveVariable | |
| CollectiveVariable () | |
| Constructor. | |
| double | GetValue () const |
| Get current value of the CV. More... | |
| virtual double | GetMinimumImage (double) const |
| Returns the minimum image of a CV based on the input location. More... | |
| virtual double | GetPeriodicValue (double location) const |
| Apply periodic boundaries to a given value. More... | |
| const std::vector< Vector3 > & | GetGradient () const |
| Get current gradient of the CV. More... | |
| const Matrix3 & | GetBoxGradient () const |
| Get gradient contribution to box. | |
| const std::array< double, 2 > & | GetBoundaries () |
| Get CV boundaries. More... | |
| virtual double | GetDifference (double location) const |
Static Public Member Functions | |
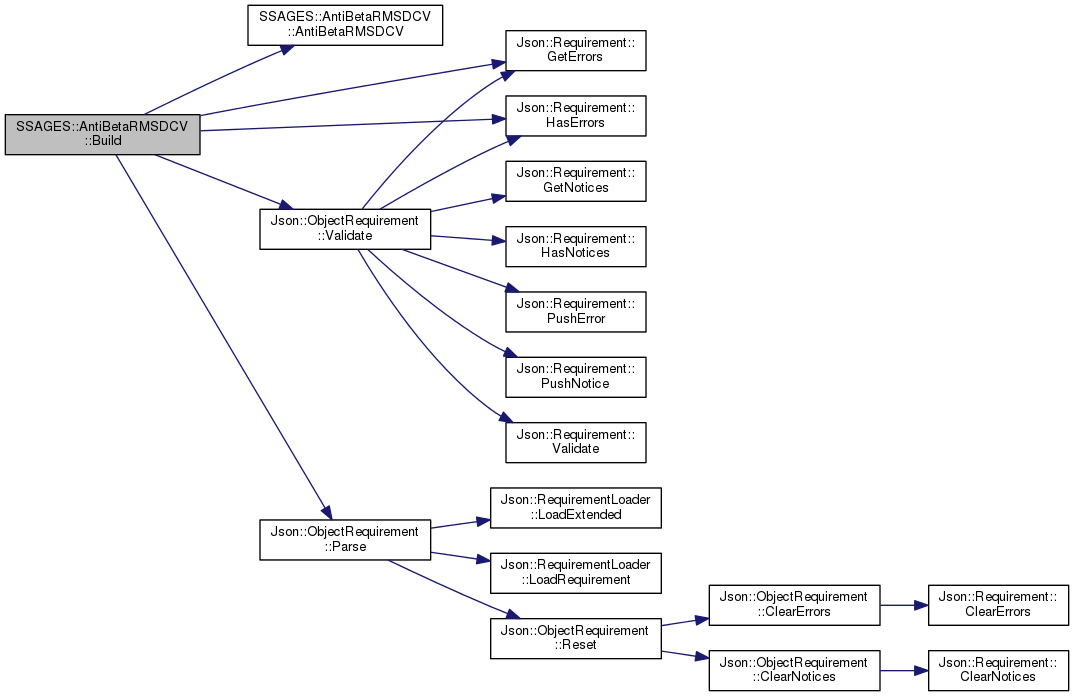
| static AntiBetaRMSDCV * | Build (const Json::Value &json, const std::string &path) |
| Set up collective variable. More... | |
| Static Public Member Functions inherited from SSAGES::CollectiveVariable | |
| static CollectiveVariable * | BuildCV (const Json::Value &json, const std::string &path) |
| Set up collective variable. More... | |
Private Attributes | |
| std::vector< int > | resids_ |
| Residue IDs for secondary structure calculation. | |
|
std::vector< std::vector < std::string > > | atomids_ |
| Atom IDs for secondary structure calculation: backbone of resids_. | |
| std::string | refpdb_ |
| Name of pdb reference for system. | |
| std::vector< Vector3 > | refalpha_ |
| Coordinates for reference structure. | |
| double | unitconv_ |
| Length unit conversion: convert 1 nm to your internal MD units (ex. if using angstroms use 10) | |
| int | mode_ |
| Specify whether to calculate beta sheet character in intra or inter mode: 0 for either, 1 for inter, 2 for intra. | |
Additional Inherited Members | |
| Protected Attributes inherited from SSAGES::CollectiveVariable | |
| std::vector< Vector3 > | grad_ |
| Gradient vector dCv/dxi. | |
| Matrix3 | boxgrad_ |
| Gradient w.r.t box vectors dCv/dHij. | |
| double | val_ |
| Current value of CV. | |
| std::array< double, 2 > | bounds_ |
| Bounds on CV. | |
Collective variable to measure antiparallel beta secondary structure.
Following treatment in Pietrucci and Laio, "A Collective Variable for the Efficient Exploration of Protein Beta-Sheet Structures: Application to SH3 and GB1", JCTC, 2009, 5(9): 2197-2201.
Check 2 blocks of 3 consecutive protein residues for RMSD from reference "ideal" antiparallel beta sheet structure.
Definition at line 43 of file AntiBetaRMSDCV.h.
|
inline |
Constructor.
| resids | IDs of residues for calculating secondary structure |
| refpdb | String of pdb filename with atom and residue indices. |
| unitconv | Conversion for internal MD length unit: 1 nm is equal to unitconv internal units |
| mode | Specification for inter/intra mode for beta sheet character |
Construct an AntibetaRMSD CV – calculates anti-beta sheet character by summing pairwise RMSD to an ideal anti-beta sheet structure for all possible 6 residue segments.
Definition at line 78 of file AntiBetaRMSDCV.h.
References resids_.
Referenced by Build().

|
inlinestatic |
Set up collective variable.
| json | JSON input value. |
| path | Path for JSON path specification. |
nullptr in case of unknown error.Builds a CV from a JSON node. Returns a pointer to the built cv. If an unknown error is encountered, this function will return a nullptr, but generally it will throw a BuildException on failure.
Definition at line 220 of file AntiBetaRMSDCV.h.
References AntiBetaRMSDCV(), Json::Requirement::GetErrors(), Json::Requirement::HasErrors(), Json::ObjectRequirement::Parse(), and Json::ObjectRequirement::Validate().
Referenced by SSAGES::CollectiveVariable::BuildCV().

|
inlineoverride |
Evaluate the CV.
| snapshot | Current simulation snapshot. |
Definition at line 146 of file AntiBetaRMSDCV.h.
References atomids_, SSAGES::Snapshot::GetLocalIndices(), SSAGES::Snapshot::GetNumAtoms(), SSAGES::Snapshot::GetPositions(), SSAGES::CollectiveVariable::grad_, mode_, refalpha_, resids_, and SSAGES::CollectiveVariable::val_.

|
inlineoverride |
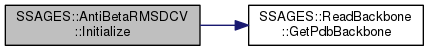
Initialize necessary variables.
| snapshot | Current simulation snapshot. |
Definition at line 104 of file AntiBetaRMSDCV.h.
References atomids_, SSAGES::ReadBackbone::GetPdbBackbone(), refalpha_, refpdb_, resids_, and unitconv_.
 1.8.7
1.8.7

